Treacher Collins Sendromu Nedir? Belirtileri Ve Tedavisi
Treacher Collins Sendromu Nedir?, Treacher Collins Sendromu Belirtileri Ve Treacher Collins Sendromu Tedavisi Hakkında merak edilenler...

KISACA TREACHER COLLİNS SENDROMU
Treacher Collins sendromu (TCS) başın ve yüzün ayırıcı özellikleriyle karakterize nadir bir genetik bozukluktur. Kraniofasial anormallikler, zigomatik kompleksin, elmacık kemiklerinin, çenelerin, damak ve ağız boşluğunun solunum yollarına ve beslenme güçlüğüne neden olabileceği azgelişmiş olma eğilimindedir. Buna ek olarak, etkilenen bireyler, üst ve alt göz kapakları (palpebral fissürler) arasındaki açılımın aşağıya eğik olması ve işitme kaybıyla sonuçlanabilecek dış ve orta kulak yapılarının anormallikleri de dahil olmak üzere gözlerin malum olmasına (oküler) sahip olabilirler. Durumun bir parçası olarak, mikrosefali ve psikomotor gecikme gibi beyin ve davranışsal anormallikler de ara sıra görülmüştür.
Treacher Collins sendromu ile ilişkili spesifik belirtiler ve fiziksel özellikler, bir bireyden diğerine büyük ölçüde değişebilir. Bazı kişiler tanı koyulamayacak kadar hafif etkilenebilirken bazıları ciddi, hayatı tehdit eden komplikasyonlar geliştirebilir.
Treacher Collins sendromu, TCOF1, POLR1C veya POLR1D genlerinde bir mutasyona neden olur. TCOF1 durumunda, kalıtım modeli otozomal dominanttır, oysa POLR1C için otozomal resesif olur. Tersine, POLR1D’deki hem otozomal dominant hem de resesif geçişli mutasyonlar Treacher Collins sendromu ile bağlantılı olarak bildirilmiştir.
Treacher Collins sendromu, 1900 yılında tıbbi literatürdeki bozukluğu ilk kez anlatan bir Londra oftalmolog Edward Treacher Collins’in adını almıştır. TCS aynı zamanda mandibulofasyal disostoz ya da Treacher Collins Franceschetti sendromu olarak da bilinir.

TREACHER COLLİNS SENDROMU BELİRTİLERİ
Treacher Collins sendromu belirtileri, bir ailenin üyeleri arasında bile bir insandan diğerine dramatik olarak değişebilir. Bazı kişiler hafiften etkilenebilir ve tanı konulamayabilir; Diğerlerinde ciddi anormallikler ve hayatı tehdit eden solunum komplikasyonları yapma potansiyeli olabilir. Etkilenen bireylerin aşağıda tartışılan belirtilerin hepsine sahip olmayacaklarını belirtmek önemlidir.
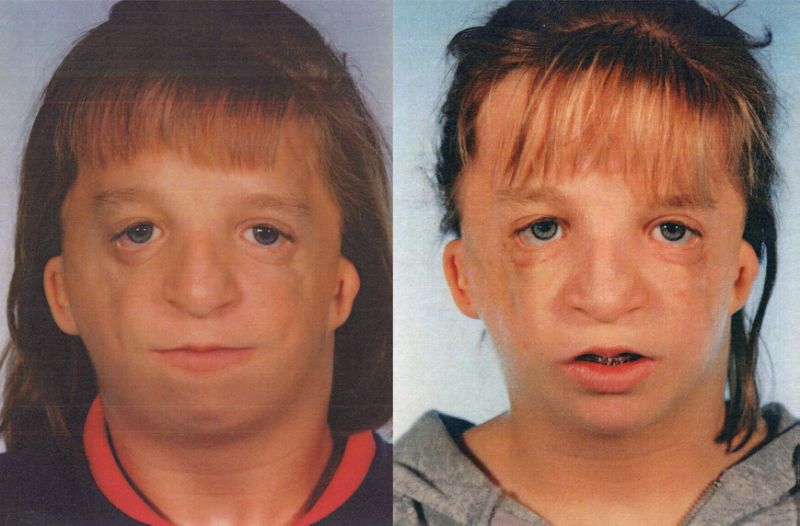
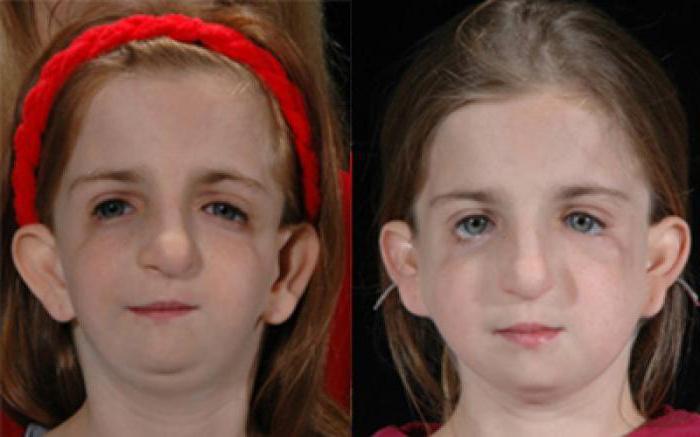
Treacher Collins sendromu’nun en belirgin belirtileri, göz çevresindeki yüz, kulaklar ve yumuşak dokuların belirli kemiklerini kapsar. Etkilenen kişiler yüz özellikleri ile kendini gösterir ve işitme ve görme problemleri gelişebilir. TCS’nin anormallikleri tipik olarak simetriktir (yüzün her iki tarafında hemen hemen aynıdır) ve doğumda (konjenital) bulunur. Konuşma ve dil gelişimi işitme kaybı, yarık damak, çene ve hava yolu sorunları ile engellenebilir. İstihbarat genellikle etkilenmez ancak durumun bir parçası olarak nadiren mikrosefali ve bilişsel gecikme gibi beyin ve davranışsal anormallikler bildirilmiştir.
TCS’li bebekler, az gelişmiş (hipoplastik) veya elmacık kemiklerine (malarlar) sahip olup, yüzün bu alanının düz veya batmış olmasına neden olur. Alt çene (mandibuler) kemiği tamamen gelişmemiş (mandibular hypoplasia), çene ve alt çenenin anormal küçüklüğüne neden olur (mikrognati). Alt çene kemiğinin kısımlarına kemiğe bağlanan belirli kemik yapıları (örneğin, koronoid ve kondiloid işlemleri) alışılmadık derecede düz veya mevcut olmayabilir. Etkilenen bebekler boğazın az gelişmesini de gösterebilirler (faringeal hipoplazi). Alt çene az gelişmiş (mandibular hypoplasia) ve çenenin anormal küçüklüğü (mikrognati) ile birlikte olan pharyngeal hipoplazi, erken bebeklik döneminde beslenme problemlerine ve solunum yetmezliklerine (solunum yetmezliği) katkıda bulunabilir. Çocuklar normal solunum ve uyku esnasında hava hareketinin defalarca tekrarlanmasıyla karakterize edilen obstrüktif uyku apnesi yaşayabilir. Ciddi derecede etkilenen bazı kişilerde, hayatı tehdit eden solunum zorlukları gelişebilir.

Solunum yollarında veya beslenme güçlüğüne katkıda bulunabilecek ek anormallikler arasında burun hava yollarının daralması veya tıkanması (choanal stenoz veya atrezi) bulunur. Bazı çocuklar ağız çatısının (yarık damak) tamamlanmamış şekilde kapatılmasına ya da tıkanmamasına rağmen ağızda normalden daha geride (glossoptoz) bulunan ciddi mikrognati içeren “Pierre Robin Sendromu” özelliklerine sahip olarak tanımlanabilir. Buna ek olarak, ağız ve çenenin malformasyonları, dişlerin az gelişmiş (hipoplastik) ve / veya yanlış hizalanmış (kötü kapanmış) diş anormalliklerine neden olabilir. Diş eksikliği (diş agenezi), diş minesinin bulanıklığı veya renk değişimi (emaye opaklığı) ve bazı üst dişlerin (maksiller molar dişlerin) uygun olmayan ektopik patlamaları da dahil olmak üzere ek dişhekimliği anormallikleri bildirilmiştir. TCS’li bireyler işitme kaybına neden olabilirler. Ses dalgalarının orta kulak yoluyla iletilmemesi (iletken işitme kaybı). İletken işitme kaybı genellikle orta kulaktaki yapıları etkileyen anormalliklerden kaynaklanır ve TCS’li bireyler, orta kulakta ses dalgalarının iletildiği üç küçük kemiğe (yani incus, malleus ve stapes) sahip yanlış veya yanlış ossiküllere de sahip olabilirler. Ek olarak, dış kulak yapıları dış kulak yollarının daralması (tıkanıklığı) veya tıkanıklığı (atrezisi) olan, genellikle eksik, küçük veya yanlış biçimlendirilmiş (mikrotia) şekildedir. Dış kulaklar buruşmuş veya döndürülebilir. Bununla birlikte, iç kulaktaki koksal spiral organın malformasyonu (koklea) ve iç kulaktaki dengede (vestibüler cihaz) rol oynayan yapılar bildirilmiş olsa da, iç kulak genellikle etkilenmez. Ek belirtiler, dış kulağın hemen önündeki cilt ya da çukurların küçük büyümelerinin (preauriküler etiketler) varlığı ve normalde kulakları burna sızdıran bir uçta (kör fistül) kapalı anormal bir geçişi içerebilir.
Treacher Collins sendromlı birçok bebeğin gözleri çevreleyen doku anormallikleri vardır. Bu göz farklılıkları, etkilenen kişilere üzgün bir yüz görünümü verebilir. En sık görülen göz semptomu, üst ve alt göz kapakları arasındaki açılma için aşağı eğimdir (palpebral fissürler). Ek belirtiler arasında, alt kapak çentikleri veya eksik kapak dokusunun yarısı (kapak kolobomu), alt göz kapağında kısmi kirpik yokluğu, çarpı gözler (şaşılık) ve gözyaşı kanallarının daralması (dacrostenoz) bulunur. Bazen dünyanın kusurları görülür ve irisin eksik dokusunun çentik veya yarık ya da anormal derecede küçük gözler (mikroftalmi) içerebilir. Bazı hastalarda görme kaybı meydana gelebilir. Görme bozukluğu derecesi, oküler anormalliklerin şiddetine ve kombinasyonuna bağlı olarak değişir. Alt göz kapağı anormallikleri gözlerin kurumasına neden olabilir, bu da kronik tahriş ve göz enfeksiyonu riskini arttırır.
Treacher Collins sendromulu bazı kişiler, geniş göz aralıkları, üst göz kapağının çentiklenmesi, burun deformitesi, anormal derecede geniş bir ağız (makro stomia), ağızda yüksek derecede kemerli bir çatı (damak), kafa derisi saçının olağandışı büyümesi gibi ek fiziksel anormallikler sergiler Yanaklara doğru, ve / veya doğuştan kalp kusuruna.
TCS’li bireylerin yaklaşık% 5’inde gelişme eksiklikleri veya psikomotor gecikme gibi nörolojik problemler görülür. Bununla birlikte, istihbarat genellikle normal dil gelişiminden etkilenmez. Bununla birlikte, işitme kaybı, yarık damak ya da yapısal bozulma nedeniyle ses üretme güçlüğü nedeniyle konuşma geliştirme ile ilgili sorunlar ortaya çıkabilir.
TREACHER COLLİNS SENDROMU NEDENLERİ
Treacher Collins sendromu, TCOF1, POLR1C veya POLR1D genlerinin mutasyonundan kaynaklanır. TCOF1 durumunda, kalıtım otozomal dominanttır, POLR1C için otozomal resesifdir ve POLR1D için otozomal dominant veya otozomal resesif olabilir.
Genetik hastalıklar, babadan ve anneden gelen kromozomlar üzerinde bulunan belli bir özellik için gen kombinasyonu ile belirlenir. Dominant genetik bozukluklar, hastalığın ortaya çıkışı için yalnızca bir adet anormal gen kopyası gerektiğinde ortaya çıkar. TCOF1 ve POLR1D için, anormal gen her iki ebeveynden de miras alınabilir veya etkilenen bireyde yeni bir mutasyonun (gen değişimi) bir sonucu olabilir. TCS olgularının yaklaşık% 60’ında mutasyon, bozukluğun önceden bir aile geçmişi olmaksızın rasgele ortaya çıkan yeni bir mutasyon (de novo mutasyon). Bununla birlikte, bir ebeveyn hafif derecede etkilenmiş olabilir ve bozukluğa sahip olduklarından habersizdir. Etkilenen ebeveynden yavruya anormal gen geçirme riski, sonuçta ortaya çıkan çocuğun cinsiyetine bakılmaksızın veya önceki gebeliklerin etkilenen veya gerçekten etkilenmeyen çocuklarla sonuçlanıp sonuçlanmadığına bakılmaksızın, her gebelik için% 50’dir.
Reseptif genetik bozukluklar (örn., POLR1C veya POLR1D mutasyonlarının neden olduğu TCS), bir birey, her ebeveynden aynı özellik için aynı anormal geni miras aldığında ortaya çıkar. Bir birey hastalık için bir normal gen ve bir gen alırsa, kişi hastalık için bir taşıyıcı olur, ancak genellikle semptom göstermez. İki taşıyıcı ebeveynin de kusurlu geni geçme riski vardır ve bu nedenle etkilenen bir çocuğa sahip olmak her gebeliğin% 25’idir. Her gebelikte ebeveyn gibi taşıyıcı olan bir çocuğa sahip olma riski% 50’dir. Bir çocuğun her iki ebeveynden normal genler alması ve bu özellik için genetik olarak etkilenmeme şansı% 25’dir.

Araştırmacılar, kromozom 5’in (5q32) uzun kolunda bulunan TCOF1 geninin mutasyonlarının TCS vakalarının çoğuna (yaklaşık% 80) neden olduğunu saptadı. İnsan hücrelerinin çekirdeğinde bulunan kromozomlar, her bir birey için genetik bilgiyi taşırlar. İnsan vücudu hücreleri normalde 46 kromozoma sahiptir. İnsan kromozomlarının çiftleri 1 ile 22 arasında numaralandırılmış ve cinsiyet kromozomları X ve Y olarak adlandırılmıştır. Erkekler bir X ve bir Y kromozomuna, kadınların iki X kromozomuna sahipler. Her kromozomda “p” ile gösterilen kısa kol ve “q” ile gösterilen uzun kol bulunur. Kromozomlar daha da numaralandırılmış birçok gruba bölünür. Örneğin, “kromozom 5q32”, kromozom 5’in uzun kolundaki 32 bandını belirtir. Numaralı bantlar, her kromozom üzerinde bulunan binlerce genin yerini belirtir.
TCOF1, treacle olarak bilinen bir proteini kodlayan (oluşturan) talimatları taşır. TİS’in gelişiminde treklerin oynadığı kesin rol tam olarak anlaşılamamıştır. Araştırmacılar, proteinlerin toplandığı hücrelerde (ribozomlar) bulunan bazı küçük yapıların yaratılışında mangalezinin rol oynadığını tespit ettiler. Bu özellikle, embriyonik gelişme sırasında çok erken oluşan ve yüzün altında yatan kemik ve kıkırdakların çoğunu oluşturan nöral krest hücreleri adı verilen bir hücre grubu oluşturulması için özellikle önemlidir. Ribozomların oluşumundaki bozukluklardan (biyogenez) kaynaklanan koşullara ribozomopati denir. RNA polimeraz I ve III’ün POLR1C ve POLR1D kodlu alt birimleri ayrıca ribozom biyogenezi için de gereklidir. TCOF1, POLR1C ve POLR1D’deki mutasyonların yetersiz protein montajına neden olduğu ve embriyonun gelişiminde hücrelerin çoğalma ve büyüme gereksinimlerini karşılamasına izin vermediği muhtemel gibi görünüyor. TCS çok değişkendir, araştırmacılar, ek genetik ve muhtemelen çevresel faktörlerin bozukluğun değişken şiddetinde rol oynayabileceğini düşünüyorlar. Bu konsepti desteklemek için son deneysel veriler, treacle’in nöral hücrelerdeki oksidatif strese bağlı DNA hasarına karşı koruma ve sinir hücresi bölünmesi sırasında spindle yönlendirmede kritik bir role sahip olduğunu ve her ikisinin daha sonra baş ve yüz gelişimini etkilediğini gösterdi.
TCS’nin özellikleri ve fizik muayene bulguları ile bazı kişiler (yaklaşık% 10-15), yukarıda bahsedilen üç genin herhangi birinde mutasyona sahip değildir; bu durum bazı durumlarda ek olarak henüz tanımlanamayan genlerin TCS’ye neden olabileceğini düşündürmektedir.
TREACHER COLLİNS SENDROMU KİMLERDE GÖRÜLÜR?
Treacher Collins sendromu erkekleri ve kadınları eşit sayıda etkiler. Genel popülasyonda yaygınlığın 10.000-50.000 kişi arasında 1 olduğu tahmin edilmektedir. Bazı hafif etkilenmiş kişiler tanı konulamayabilir, bu da genel popülasyonda bozukluğun gerçek sıklığının saptanmasını zorlaştırır.
TREACHER COLLİNS SENDROMU İLE İLİŞKİLİ RAHATSIZLIKLAR
Aşağıdaki rahatsızlıkların belirtileri Treacher Collins sendromuna benzer olabilir. Karşılaştırmalar ayırıcı tanıda yararlı olabilir.
Akrofasiyal disostoz, mandibulofasyal disostozis koşullarında (yani TCS) gözlenen kraniofasiyal anomalilere benzer konjenital sendromları, ancak ekstremite defekti eklenerek tanımlanmaktadır. Bunlar, iyi karakterize Nager sendromu ve Miller sendromu bozukluklarının yanı sıra, akrofasyal disostoz, Cincinnati tipi gibi daha yakın zamanda tanımlanan durumları içerir.
Nager sendromu (aynı zamanda akrofasyal disostoz olarak bilinir – Treacher Collins tipi ekstremite anomalileri), kollar, eller ve / veya ayaklardaki anormalliklerle birlikte görülen TCS’deki kraniofasiyal malformasyonlar ile karakterize nadir bir kalıtsal bozukluktur. Kraniofasial malformasyonlar, elmacık kemiklerinde az gelişmişlik (malar hipoplazi); Alt çene (mandibular hypoplasia) gelişimi eksik, çene anormal derecede küçük görünmesine neden olur (mikrognati); Işitme kaybına (iletken işitme kaybı) neden olan, hipoplastik ve bozuk biçimli (displastik) dış kulaklar (pinnae) ve kör biten veya yoksun dış kulak kanalları (mikrotia); Ve aşağıya eğik palpebral fissürler, alt kirpiklerin yokluğu veya yokluğu ve sulanan üst göz kapaklarının (pitoz) tedavisi. Bacaklarda anormallikler az gelişmişlik veya baş parmaklarının yokluğu, önkoldaki kemiklerin birinin olmaması (radius), kemiklerin önkoldaki anormal füzyonu (radioulnar synostosis), bazı parmakların sürekli fleksiyonu (kamptodaktili) ve Ayak sesleri (sindaktili). Nager Sendromu tipik olarak kromozom 1q12-q21 üzerindeki SF3B4 geninde heterozigot mutasyon yoluyla otozomal dominant özellik nedeni olarak miras alınır. Çoğu vakada, Nager sendromu ailede rastgele ve yeni ortaya çıkar (sporadik) ancak otozomal resesif kalıtım daha önce tanımlanmıştır.
Miller sendromu (postaksiyel akrofasyal disostoz olarak da bilinir) kollar, eller ve ayaklardaki anormalliklerle birlikte görülen kraniofasiyal malformasyonlar ile karakterize nadir bir kalıtsal bozukluktur. Kraniofasial malformasyonlar, elmacık kemiklerinde az gelişmişlik (malar hipoplazi); Anormal derecede küçük bir alt çene (mikrognati); Ağız çatısının tamamlanmamış kapatılması (yarık damak); Küçük, çıkıntılı, “bardak biçimli” kulaklar; Ve alt gözkapaklarından doku yokluğu (kolobomlar). Ekstremite anormallikleri, eksik gelişimi (hipoplazi), ağlamayı (sindaktiliğini) ve belirli parmakların veya ayak parmaklarının yokluğunu içerebilir; Bazı parmakların uygun olmayan şekilde konumlandırılması; Ve önkollarda kemiklerin anormal füzyonu (radioulnar synostosis) ve uygunsuz gelişim, ve böylece önkolların olağandışı kısa görünmesine neden olur. Bazı hastalarda ek fiziksel anormallikler görülebilir. Miller sendromu, 16q22.2 kromozomundaki DHODH geninde bileşik heterozigot mutasyonların neden olduğu otozomal resesif bir özellik olarak kalıtılır.
Acrofacial dysostosis, Cincinnati tipi, pelvik ve basamak anomalileri içeren ekstremite iskelet kusurlarıyla birlikte veya eklemsiz olarak gelişmemiş zigomatik kemerler (malar hipoplazi), maksilla ve mandibula (mikrognati) ile karakterize nadir bir kalıtsal bozukluktur. Treacher Collins sendromuna benzer şekilde, etkilenen bireylerin değişken fenotipleri vardır ve ek eğik özellikler arasında aşağı eğik göz yarıkları (palpebral fissürler), göz kapaklarının yarıkları (koloboma), hatalı şekillendirilmiş (choanal atrezi) veya yokluğunda burun (anoti) ve iletken işitme kaybı bulunur. Limb ve pelvik anomaliler arasında kısa ve geniş rakamlar, kısa eğilmiş femurlar, kemik oluşumunda gecikmeli veya var olmayan (ossifikasyon) sayılabilir. Bugüne kadar, etkilenen tüm bireylerin, RNA Polimeraz 1’in en büyük alt birimini kodlayan kromozom 2q11.2 üzerindeki POLR1A’da bir heterozigot mutasyona sahip oldukları bulundu. POLR1A, aynı zamanda ribozomal DNA genlerini (rDNA) transkribe eden RNA Polimeraz 1’in katalitik altbirimi idi. Ribozomal RNA (rRNA) öncüllerine dönüştürür. Bu süreç ribozom biyojenezinin hız sınırlayıcı adımlarından biridir.
Hemifasiyal mikrozomi, çene, ağız ve kulak içeren kraniofasial anormalliklerle karakterize, kalp, iskelet, böbrek sistemleri ve ekstremitelerin ekstra kraniyal anomalileri (genişlemiş spektrumlu HFM) ile karakterize nadir bir hastalıktır. Birçok araştırmacı Goldenhar sendromunu hemifasiyal mikrozominin bir varyantı ve alt grubu olarak düşünmektedir. Tıbbi literatürde, hemifasiyal mikrozomi ve Goldenhar sendromu, genellikle “Göz Küresi Übertebral (OAV) Spektrumu” veya “kraniofasiyal mikrozomi” terimi altında gruplandırılır. Çoğu vakanın belirgin bir nedeni olmaksızın (dağınık) rasgele ortaya çıkar. Bazı vakalarda, bazı araştırmacılara göre, otozomal dominatif kalıtım öneren pozitif bir aile geçmişi vardır. Kraniofasial mikrozom ile ilişkili fiziksel özellikler kişiden kişiye dramatik şekilde değişir. Bu özellikler vücudun bir tarafını içerir (tek taraflı) ve belirli anormalliklerin çeşitli kombinasyonlarını temsil edebilir. Bunlar, elmacık kemiklerinde, üst çenede ve alt çenede (malar, maksiller ve mandibular hipoplazi) az gelişme; Yüzdeki belli kasların az gelişmesi; Dildeki anormallikler, ağız çatısının (yarık damak) tamamlanmamış şekilde kapanması ve / veya dudakta anormal bir oluk (yarık dudak); Kör uçlu veya harici kulak kanalları olmayan (mikrotiya) hatalı biçimlendirilmiş dış kulaklar (pinna), işitme kaybına (iletken işitme kaybı) neden olur; Kulaklardaki deride anormal gelişmeler (cilt etiketleri); Ve / veya spinal kolondaki bazı kemiklerin (vertebral hipoplazi) eksik oluşu. Ek anomaliler, üst göz kapaklarından, gözlerin çaprazlandığı (şaşılık) ve / veya anormal derecede küçük gözlerden (mikroftalmi) oluşan dokunun kısmen veya tamamen yokluğunu (kolobom); Kalp (kalp) defektleri; Böbrek (böbrek) anormallikleri; Ve / veya ek fiziksel anormallikler. TCS’yi OAV Spektrumundan ayıran iki önemli unsur şunlardır: 1) TCS simetrik; Ve 2) TCS sinirleri etkilemez.
TREACHER COLLİNS SENDROMU TEŞHİSİ
Treacher Collins sendromu’nun teşhisi kapsamlı bir klinik değerlendirmeye, detaylı bir hasta öyküsüne ve karakteristik fiziksel bulguları tanımaya dayanmaktadır. Malformasyon veya dış kulak yokluğu gibi bir çok ilişkili anormallikler doğumda (konjenital) görülür.
TREACHER COLLİNS SENDROMU İLE İLGİLİ KLİNİK VE TEST ÇALIŞMALARI
Uzmanlaştırılmış röntgen çalışmaları, belli gözlenen kraniyofasial anormalliklerin varlığını ve / veya kapsamını teyit edecektir. Örneğin, bu tür görüntüleme testleri, alt çene kemiğinin az gelişmiş olması (mandibular hipoplazi), kafatasının belirli bölümlerini etkileyen hipoplazinin varlığı ve / veya miktarı ve / veya varlığın varlığı nedeniyle çenenin anormal küçüklüğünü (mikrognati) gösterir Klinik değerlendirme sırasında görülemeyen kulağın ek malformasyonları.
Buna ek olarak, az sayıda belirtiyi sergileyen etkilenmiş bireylerde, kraniyofasiyal bölgenin ayrıntılı klinik muayenesi ve X ışını görüntülemesi, TCS ile bağlantılı bazı karakteristik özelliklerin (örn., Zigomatik kemerlerin hipoplazisi) varlığını ince gösterebilir. TCS, diğer kraniofasial sendromlarda ortaya çıkabilecek birkaç fiziksel özelliği paylaştığından, birçok araştırmacı, teşhis doğrulamasının moleküler genetik test yoluyla ve / veya bazı durumlarda dikkatli ve detaylı bir aile geçmişi ile yapılmasını önermektedir.
Teşhisi doğrulamak için moleküler genetik test, TCOF1, POLR1C ve POLR1D genlerindeki mutasyonları saptamak için ticari ve akademik araştırma laboratuvarlarında mevcuttur. Bireylerin yaklaşık% 90-95’inde TCOF1 geninin tanımlanabilir bir mutasyonu vardır. Ayrıca, bir TCOF1, POLR1C veya POLR1D mutasyonunun genetik olarak doğrulanması, etkilenen bir aile üyesinde bir mutasyon saptanması durumunda, doğumdan önce (prenatal olarak) amniyosentez ve koriyonik villüs örneklemesi ile saptanabilir. Bazı durumlarda, gelişen fetüsün bir görüntüsü oluşturmak için yansıyan ses dalgalarını kullanan fetal ultrasonografi, TCS’yi düşündüren karakteristik bulguları ortaya çıkarabilir. TCS tanısı konan bir kişinin akrabaları, özellikle ebeveynler ve kardeşler dikkatli bir şekilde incelenmelidir, çünkü hafif olgular genellikle fark edilmeden tanı konulmaz ve tanı konulmaz.
TREACHER COLLİNS SENDROMU TEDAVİSİ
Treacher Collins sendromu için herhangi bir tedavi yoktur. Tedavi her bireyde belirgin olan spesifik semptomlara yöneliktir. Tedavi bir uzman ekibinin eşgüdümlü çabalarını gerektirebilir. Pediatric, pediyatrik kulak, burun-boğaz uzmanları (pediyatrik kulak Burun boğaz uzmanı), pedodontik diş hekimi, pediyatri hemşiresi, plastik cerrah, konuşma patologları, odalologlar, oftalmologlar, psikologlar, genetikçiler ve diğer sağlık profesyonelleri, bir çocuğun tedavisini sistematik ve kapsamlı bir şekilde planlamaya ihtiyaç duyabilirler.
Doktorlar, bozuklukla ilişkili olabilecek bazı anormallikleri tespit etmek için TCS’li bireyleri düzenli olarak izlemektedir. Örneğin, işitme kaybının herhangi bir başlangıcını algılamak için etkilenen bir kişinin işitmesi dikkatle izlenmelidir. Bebeğin işitme değerlendirmesi kritik önem taşıyor. Doğru bir konuşma gelişimini sağlamak için, tam bir değerlendirme, bir yaşından önce ve daha sonra yılda bile olsa, yaşam boyunca erkenden yapılmalıdır.

Herhangi bir görme bozukluğu olasılığını tespit etmek için gözün iç kısmını görselleştirmek için bir araç (oftalmoskop) kullanılır. Bu muayene, TCS ile bağlantılı gözlerin anormalliklerini sergileyen (örn., Kolobomalar, şaşılık, mikroftalmiler) uygun önleyici adımları ve / veya tedaviyi hızlandırmak için önemlidir. Etkilenen kişiler ayrıca çene ve dişhekimliği anormallikleri açısından izlenmelidir.
Etkilenen çocukların potansiyellerine ulaşmalarını sağlamak için erken müdahale önemlidir. Yararlı olabilecek özel hizmetler konuşma terapisi, özel sosyal destek ve diğer tıbbi, sosyal ve / veya mesleki hizmetleri içerebilir. Genetik danışma, etkilenen kişiler ve aileleri için fayda sağlayacaktır.
TREACHER COLLİNS SENDROMU AMELİYATI
Bazı hastalarda, kraniyofasiyal malformasyonların cerrahi rekonstrüksiyonu gerekli olabilir. Yarık damak onarımı, çene yeniden yapılandırma veya kafatasındaki diğer kemiklerin onarımı (örneğin, malar kemikler, zigomatik kompleks) için ameliyat yapılabilir. Kullanılan spesifik cerrahi prosedürler ve ameliyat yapıldığı yaş, malformasyonların ciddiyetine, genel sağlık durumuna ve kişisel tercihine bağlıdır.
Örneğin, farklı anormallikler farklı yaşlarda tedavi edilebilir. Yarık damak sıklıkla 1-2 yaş civarında düzeltilir. Zigomatik ve yörünge rekonstrüksiyonu genellikle 5-7 yaşlarında görülür. Dış ve iç kulak rekonstrüksiyonu genellikle 6 yaş civarında gerçekleşir. Çene kemiğinin uzatılması veya yeniden yapılandırılması, durumun derecesine ve şiddetine bağlı olarak yenidoğandan ergen yıla değişebilir.
Obstrüktif hava yolları ebeveynlere veya klinisyenlere her zaman açıkça görülen ciddi bir sorun olabilir. Tıkanıklığın şiddetinin belirlenmesine yardımcı olmak için uyku ya da uyku çalışması yapılabilir ve tedavi planını etkileyebilir. Ciddi derecede etkilenen bireylerde, trakeostomi olarak adlandırılan ve etkili bir hava yolu korumak için nefes borusuna (trakea) cerrahi olarak bir tüp yerleştirilebilir. Mandibular distraksiyon olarak bilinen, çene kemiğinin uzunluğunu arttırmak için kullanılan bir prosedür gerekli olabilir. Yeme zorlukları yaşayan etkilenen bebeklerin yeterli miktarda kalori (gastrostomi) aldığından emin olmak için mideye ameliyatla bir tüp yerleştirilebilir.
TCS ile potansiyel olarak ilişkili çeşitli kranyofasiyal anomalileri tedavi etmek için birden fazla ameliyat yapılması gerekebilir. Ameliyat sayısına rağmen sonuçlar bir kişiden kişiye değişir ve nihai sonuç nadiren tamamen düzeltilebilir.
Bazı bireylerde, orta kulak malformasyonlarının ve buna bağlı iletken işitme kaybının düzeltilmesi için bir operasyon yapılabilir. Bununla birlikte, çoğu hastada, kemiğe bağlı işitme cihazları (BAHA) gibi özel işitme cihazları, ameliyattan ziyade yeterli olabilir. Kemik bağlantılı işitme cihazları, sesi dış kulak kanalını ve orta kulağı atlamak suretiyle doğrudan kemik yoluyla iç kulağa iletir (her ikisi de TCS’li bireylerde sıklıkla etkilenir). Dış kulak malformasyonlarının fonksiyonel ve çevresel etkiler için düzeltilmesi için rekonstrüktif cerrahi yapılabilir Kozmetik nedenlerle, genellikle dış kulak rekonstrüksiyonu yapılmalıdır. (Treacher Collins Sendromlu Doğduğu İçin Annesi Tarafından Reddedilen Bebeğin Hikayesi Buradan Okuyabilirsiniz!)











En Çok Sevdiğiniz Renk Hangisi?
| İmsak | 04:01 | ||
| Güneş | 05:48 | ||
| Öğle | 13:16 | ||
| İkindi | 17:11 | ||
| Akşam | 20:33 | ||
| Yatsı | 22:12 |




| Takımlar | O | P |
|---|---|---|
| 1. Galatasaray | 38 | 102 |
| 2. Fenerbahçe | 38 | 99 |
| 3. Trabzonspor | 38 | 67 |
| 4. Başakşehir | 38 | 61 |
| 5. Kasımpasa | 38 | 56 |
| 6. Beşiktaş | 38 | 56 |
| 7. Sivasspor | 38 | 54 |
| 8. Alanyaspor | 38 | 52 |
| 9. Rizespor | 38 | 50 |
| 10. Antalyaspor | 38 | 49 |
| 11. Gaziantep FK | 38 | 44 |
| 12. A.Demirspor | 38 | 44 |
| 13. Samsunspor | 38 | 43 |
| 14. Kayserispor | 38 | 42 |
| 15. Hatayspor | 38 | 41 |
| 16. Konyaspor | 38 | 41 |
| 17. Ankaragücü | 38 | 40 |
| 18. Karagümrük | 38 | 40 |
| 19. Pendikspor | 38 | 37 |
| 20. İstanbulspor | 38 | 16 |
| Takımlar | O | P |
|---|---|---|
| 1. Eyüpspor | 34 | 75 |
| 2. Göztepe | 34 | 70 |
| 3. Sakaryaspor | 34 | 60 |
| 4. Bodrumspor | 34 | 57 |
| 5. Ahlatçı Çorum FK | 34 | 56 |
| 6. Kocaelispor | 34 | 55 |
| 7. Boluspor | 34 | 53 |
| 8. Gençlerbirliği | 34 | 51 |
| 9. Bandırmaspor | 34 | 50 |
| 10. Erzurumspor | 34 | 44 |
| 11. Ümraniye | 34 | 43 |
| 12. Manisa FK | 34 | 40 |
| 13. Keçiörengücü | 34 | 40 |
| 14. Adanaspor | 34 | 39 |
| 15. Şanlıurfaspor | 34 | 38 |
| 16. Tuzlaspor | 34 | 38 |
| 17. Altay | 34 | 10 |
| 18. Giresunspor | 34 | 7 |
| Takımlar | O | P |
|---|---|---|
| 1. M.City | 38 | 91 |
| 2. Arsenal | 38 | 89 |
| 3. Liverpool | 38 | 82 |
| 4. Aston Villa | 38 | 68 |
| 5. Tottenham | 38 | 66 |
| 6. Chelsea | 38 | 63 |
| 7. Newcastle | 38 | 60 |
| 8. M. United | 38 | 60 |
| 9. West Ham United | 38 | 52 |
| 10. Crystal Palace | 38 | 49 |
| 11. Brighton | 38 | 48 |
| 12. Bournemouth | 38 | 48 |
| 13. Fulham | 38 | 47 |
| 14. Wolves | 38 | 46 |
| 15. Everton | 38 | 40 |
| 16. Brentford | 38 | 39 |
| 17. Nottingham Forest | 38 | 32 |
| 18. Luton Town | 38 | 26 |
| 19. Burnley | 38 | 24 |
| 20. Sheffield United | 38 | 16 |
| Takımlar | O | P |
|---|---|---|
| 1. Real Madrid | 38 | 95 |
| 2. Barcelona | 38 | 85 |
| 3. Girona | 38 | 81 |
| 4. Atletico Madrid | 38 | 76 |
| 5. Athletic Bilbao | 38 | 68 |
| 6. Real Sociedad | 38 | 60 |
| 7. Real Betis | 38 | 57 |
| 8. Villarreal | 38 | 53 |
| 9. Valencia | 38 | 49 |
| 10. Deportivo Alaves | 38 | 46 |
| 11. Osasuna | 38 | 45 |
| 12. Getafe | 38 | 43 |
| 13. Celta Vigo | 38 | 41 |
| 14. Sevilla | 38 | 41 |
| 15. Mallorca | 38 | 40 |
| 16. Las Palmas | 38 | 40 |
| 17. Rayo Vallecano | 38 | 38 |
| 18. Cadiz | 38 | 33 |
| 19. Almeria | 38 | 21 |
| 20. Granada | 38 | 21 |




























